
医会在线
返回>北大科研团队发现——橙皮苷有心脏保护和抗肿瘤作用 !

心肌缺血/再灌注(I/R)损伤已成为缺血性**病的重要治疗靶点,缺血性**病是全球发病率和死亡率的主要原因。目前尚无减轻**I/R损伤的有效治疗方法。已有研究报道CaMKII (钙调素依赖性激酶II)在**疾病中起关键作用,抑制CaMKII是保护心肌损伤和预防**疾病的重要策略。但到目前为止,还没有针对CaMKII的**病治疗临床药物。此外,目前还没有选择性的CaMKII-δ抑制剂(这是**中主要的CaMKII亚型)。
近日,北京大学心血管研究所张岩团队与北京大学化学与分子工程学院雷晓光研究员以及北京大学未来技术学院肖瑞平教授合作,发现了防治缺血性**病和心力衰竭的小分子化合物—橙皮苷,研究成果以“Novel CaMKII-δ Inhibitor Hesperadin Exerts Dual Functions to Ameliorate Cardiac Ischemia/Reperfusion Injury and Inhibit Tumor Growth”为题,于2022年3月23日在Circulation(IF29.69)杂志上在线发表。
为寻找新的CaMKII抑制剂作为人类**疾病的潜在治疗药物,作者利用CaMKII-δ9(CaMKII-δ9是人类**中最丰富的CaMKII-δ剪接变体)重组蛋白建立筛选体系,初筛检测到4160个化合物具有激酶抑**用(图1A和1B)。在二次筛选中,作者根据抑制率、IC50(图1C)选择37个具有心肌保护作用的化合物进一步分析(图1D)。随后,作者采用新生大鼠的心肌细胞构建CaMKII-δ9过表达模型,用于评估化合物对细胞死亡标志物Caspase 3/7活性和LDH的抑制效果,结果发现只有hesperadin(橙皮苷,此前作为Aurora B激酶抑制剂被报道)能显著降低心肌细胞死亡,表明它可能是一个有前途的心血管化合物。

图1 基于小分子化合物库筛选CaMKII抑制剂
因CaMKII具有多种亚型,为测试Hesperadin对不同CaMKII亚型和剪接变体的选择性(结构如图2A所示),作者使用重组蛋白的CaMKII活性测定系统,分析了Hesperadin的抑**用。结果发现Hesperadin对CaMKII-α、-β、-γ和-δ(δ9)的IC50值分别为1.259、16.218、1.318和0.073 μmol/L(图2B-2E)。因此,Hesperadin对CaMKII-δ的选择性比其他亚型高约17-200倍(图2F)。接下来,作者研究了Hesperadin对**中重要的CaMKII-δ剪接变体(包括CaMKII-δ2、-δ3和-δ9)的选择性。IC50值分别为43.0、32.0和41.0 nmol/L(图2G-2I),表明Hesperadin对这些CaMKII-δ剪接变体的效力相似(图2J)。此外,作者从Protein Data Bank下载的晶体结构进行计算对接,发现Hesperadin占据了CaMKII-δ的活性位点;通过表面等离子体共振,发现Hesperadin直接与CaMKII-δ9结合,而CaMKII的自主活性被Hesperadin抑制;此外Hesperadin对CaMKII-δ9的抑制动力学是ATP竞争性的(图S3A-E)。基于这些数据,Hesperadin是一个ATP竞争性的CaMKII-δ抑制剂。
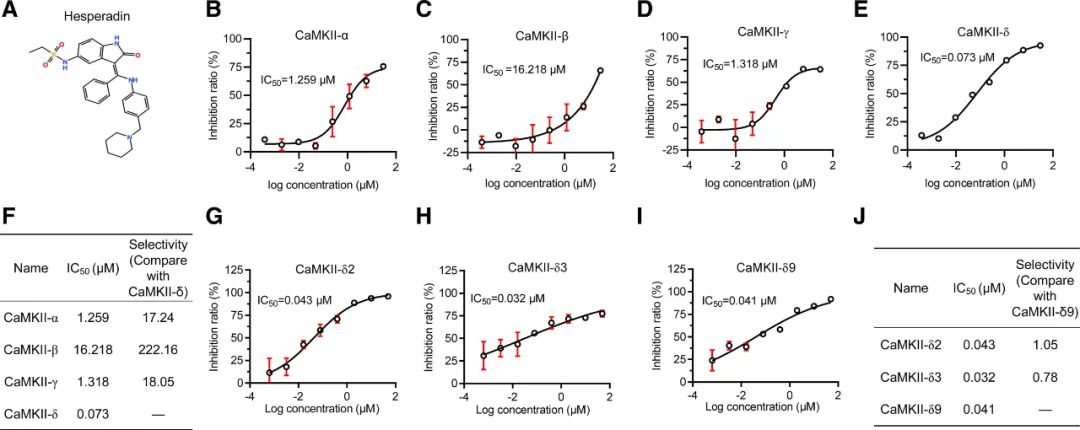
图2 Hesperadin是一种选择性CaMKII-δ抑制剂
筛选结果表明Hesperadin对小鼠心肌具有保护作用。为进一步证实这一结论,作者将Hesperadin浓度增加到0.2和0.5μmol/L,并使用caspase 3/7、LDH和caspase 3表征心肌细胞死亡。结果发现Hesperadin可剂量依赖性地减少CaMKII-δ9过表达诱导的大鼠心肌细胞死亡(图3A, B)。根据新生大鼠心肌细胞caspase 3/7活性,作者发现Hesperadin介导的保护心肌细胞死亡的IC50为0.74 μmol/L(图3C)。此外,Hesperadin显著降低了CaMKII-δ的磷酸化水平(图3D),Hesperadin对CaMKII-δ磷酸化抑制的IC50为0.21 μmol/L(图3E)。
作者最近发现CaMKII-δ9诱导心肌细胞DNA损伤(这与多种**疾病密切相关)。研究中,作者证明了Hesperadin也可以通过降低γH2AX(细胞DNA损伤的标记物)水平减轻CaMKII-δ9诱导的心肌细胞DNA损伤(图3F)。接下来,作者利用新生大鼠心肌细胞缺氧/复氧(H/R)损伤模型来模拟临床心肌I/R损伤。Hesperadin显著降低H/R诱导的细胞死亡(与caspase 3/7活性、LDH浓度和cleaved-caspase 3)、CaMKII-δ活化和心肌细胞DNA损伤(图3G-3J)。这些数据表明,Hesperadin能保护心肌细胞免受CaMKII-δ9过表达和H/R损伤引起的死亡。
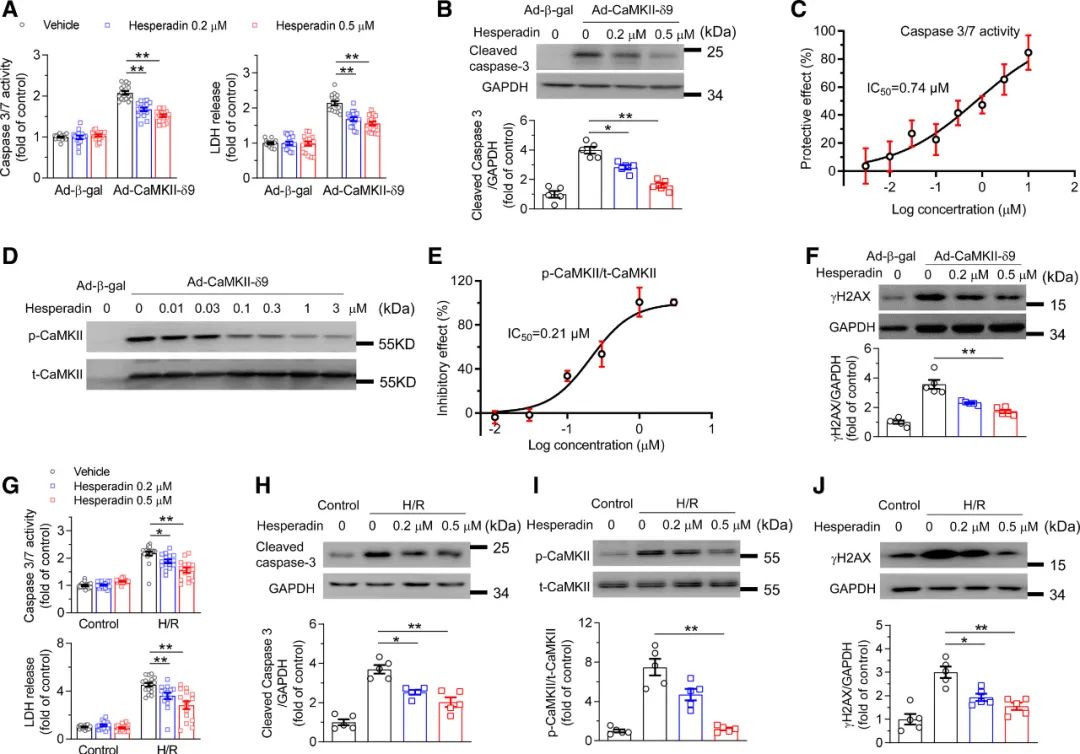
图3 Hesperadin可减轻CaMKIIδ9过表达和缺氧/复氧诱导的心肌细胞死亡
上述结果作者验证了Hesperadin在体外对心肌细胞的保护作用,那么,Hesperadin在体内是否也能发挥心肌保护作用呢?
接下来,作者在缺血30分钟(冠状动脉结扎)后再灌注24小时的小鼠急性**I/R模型中分析了Hesperadin在体内的**保护作用(图4A)。在雄性小鼠中,Hesperadin预**(缺血发生前)能显著降低I/R诱导的心肌梗死面积,从42.2±1.8%降至25.9±1.5%(图4B)。此外,Hesperadin改善了心肌细胞的死亡,主要表现是血清中LDH浓度和TUNNEL阳性细胞降低(图4C和4D)。此外,作者发现急性I/R损伤导致**内显著的DNA损伤和CaMKII激活,Hesperadin可减轻这种损伤(图4E和图S4)。雌鼠中也观察到相同的结果(图S5)。由于大多数临床患者**缺血是不可预测的,作者还检测了Hesperadin对缺血后心肌I/R损伤的保护作用(治疗后;图4F)。与前**相似,Hesperadin后**可保护**免受I/R损伤,包括心肌梗死、心肌细胞死亡和DNA损伤(图4G-4J)。
CaMKII还在**重构和心力衰竭的发展中发挥核心作用,心力衰竭是多种心血管疾病的终点。接下来,作者评估了Hesperadin在I/R诱导的心力衰竭中的作用,在30分钟**缺血后4周再灌注的小鼠心力衰竭模型中(图4K),作者发现Hesperadin减轻了心功能障碍(图4L)。此外,Hesperadin还能减少心肌细胞死亡和**慢性I/R模型的DNA损伤(图4M-4O)。
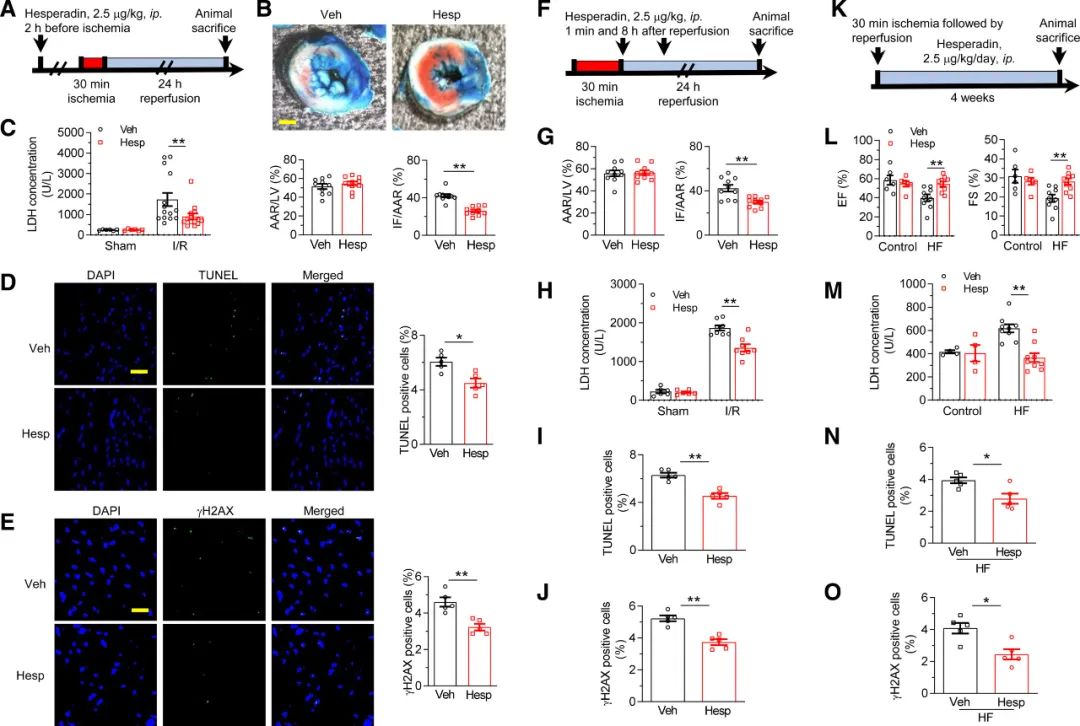
图4 Hesperadin保护**免受I/R诱导的**损伤和心力衰竭
上述的结果表明了Hesperadin在I/R诱导的**损伤和心力衰竭中具有保护作用,因CaMKII在**重构和心力衰竭的发展中发挥核心作用,因此作者进一步研究Hesperadin在**重构和心力衰竭中的作用。
作者接下来使用冠状动脉永久性结扎的心肌梗死模型(图5A),结果发现心梗诱导的心肌细胞死亡、心肌重构和心衰被Hesperadin显著改善(图5B-G)。此外,CaMKII-δ9的过表达也被证明会引起**重构和心力衰竭。因此,作者研究了Hesperadin(2.5 μg·kg-1·d-1,持续6周)在CaMKII-δ9**特异性过表达小鼠中的作用(图5H)。相似地,作者发现Hesperadin改善CaMKII-δ9诱导的**功能障碍、壁增厚、心肌重构不良、心肌细胞死亡、DNA损伤以及CaMKII活性的调节(图5I-5O)。在这些数据的基础上,Hesperadin在多个模型中减轻了**重构和心力衰竭。
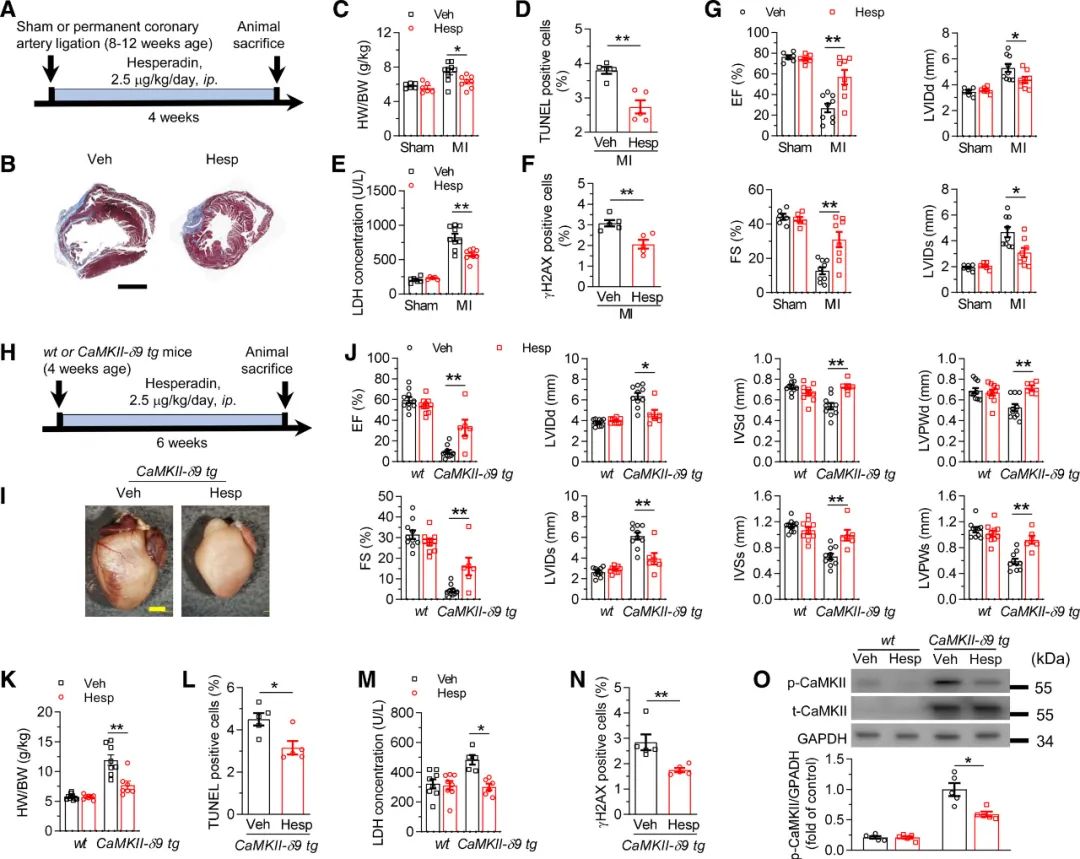
图5 Hesperadin可减轻心梗模型中的**损伤、心肌重塑和心力衰竭
上述研究结果揭示了Hesperadin过抑制CaMKII-δ发挥**保护作用,那么,CaMKII-δ缺失是否能够减少心肌损伤的发生呢?
作者研究了**CaMKII的主要亚型CaMKII-δ缺乏的小鼠模型中Hesperadin的功能。作者使用CRISPR-Cas9系统,构建了CaMKII-δ KO小鼠,经Western blots证实(图6A, B)。与野生型相比,CaMKII-δ KO小鼠中Hesperadin未能减少心肌梗死面积、TUNNEL阳性细胞、γh2ax表达,和血清LDH浓度(图6C-F)。此外,在慢性I/R诱导的心肌细胞死亡、心功能障碍和炎症细胞浸润方面,空白对照和Hesperadin组之间也没有差异(图6G-6K),这表明CaMKII-δ对于Hesperadin的急性和慢性损伤心肌保护都是必需的。
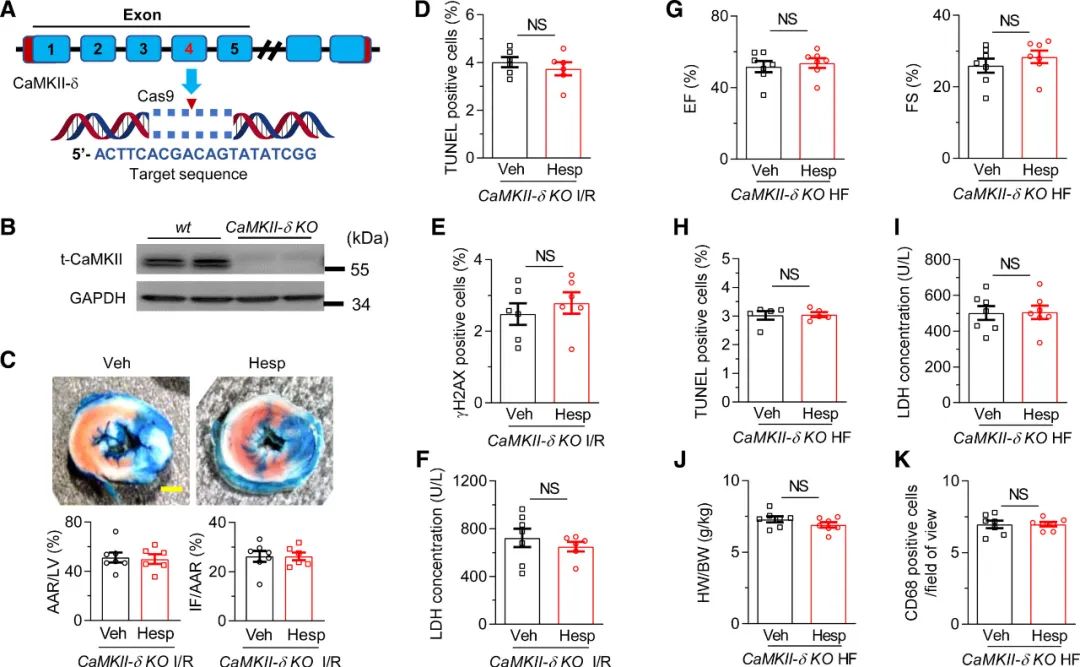
图6 CaMKII-δ在Hesperadin介导的**保护中是必需的
前面的研究结果表示,Hesperadin介导CaMKII-δ保护小鼠心肌免受损伤,那么CaMKII-δ在人**中是否能降低心肌损伤事件的发生?
已经确定的是,不同的CaMKII-δ剪接变体在**中具有不同甚至相反的作用,作者最近的研究表明,人类与啮齿动物**CaMKII-δ剪接变体的组成非常不同,表明CaMKII-δ在**中可能具有物种特异性的作用。因此,在肯定了Hesperadin对啮齿动物**的保护作用后,作者下一步研究了Hesperadin在胚胎干细胞来源的人心肌细胞中的作用。与啮齿动物心肌细胞相似,CaMKII-δ9过表达和H/R诱导的细胞死亡上调(图7A, D)、DNA损伤(图7B, E)和CaMKII-δ磷酸化(图7C, F)均被Hesperadin抑制,表明Hesperadin可以减少CaMKII激活和人心肌损伤。

图7 Hesperadin通过抑制CaMKII-δ保护人心肌细胞免受心肌损伤
Hesperadin最初被鉴定为一种抗肿瘤药物,通过抑制Aurora B激酶在体外能有效地诱导多种肿瘤细胞的生长阻滞和凋亡。本研究中作者验证了Hesperadin抑制人乳腺癌细胞系MCF-7的增殖并增加细胞死亡(图8A, B)。为进一步确定Hesperadin是否具有体内抑**用,作者将MCF-7细胞移植到BALB/c裸鼠体内,然后分别用溶剂或Hesperadin(2.5 μg·kg-1·d-1;图8 c)给药。结果显示:与对照相比,Hesperadin治疗显著延缓了肿瘤的生长(图8D)。在结束时(22天),使用Hesperadin治疗的异种移植瘤的体积更小,重量更轻(图8E),表明Hesperadin在体内外都是一种抗肿瘤药物。
值得注意的是,在裸鼠中每日使用Hesperadin治疗,12天后并没有引起心功能的改变或心肌损伤(图8F-H)。此外,在正常小鼠中,以能够改善**损伤和抑制肿瘤生长的剂量(2.5 μg·kg-1·d-1)慢性治疗(4周)没有引起任何显著的脱靶不良反应,包括体重、食物摄入、能量消耗、空间学习和记忆的异常、**和**功能、血象和主要器官的形态学等(图S6),突出Hesperadin作为一种安全的治疗心血管疾病和癌症的候选药物。
作者还检测了Hesperadin对肿瘤(MCF-7)细胞和正常分裂细胞(包括人脐静脉内皮细胞和大鼠血管平滑肌细胞)的TC50,分别为1.01、9.18和10.5 μmol/L(图S7A-S7D),说明Hesperadin对肿瘤细胞生长的抑**用是正常分裂细胞的约10倍。这可能与肿瘤细胞和正常分裂细胞中Aurora B激酶水平不同有关。此外,作者的数据显示:正常分裂的细胞中Aurora B激酶的水平远低于肿瘤细胞(图S7E),这导致了它们对Hesperadin的不同反应。关于CaMKII-δ参与Hesperadin介导的肿瘤抑制,作者发现在体内和体外Hesperadin仍然有效地抑制了缺乏CaMKII-δ的肿瘤细胞的增殖(图S8),这表明Hesperadin的抗肿瘤作用并不需要CaMKII-δ。
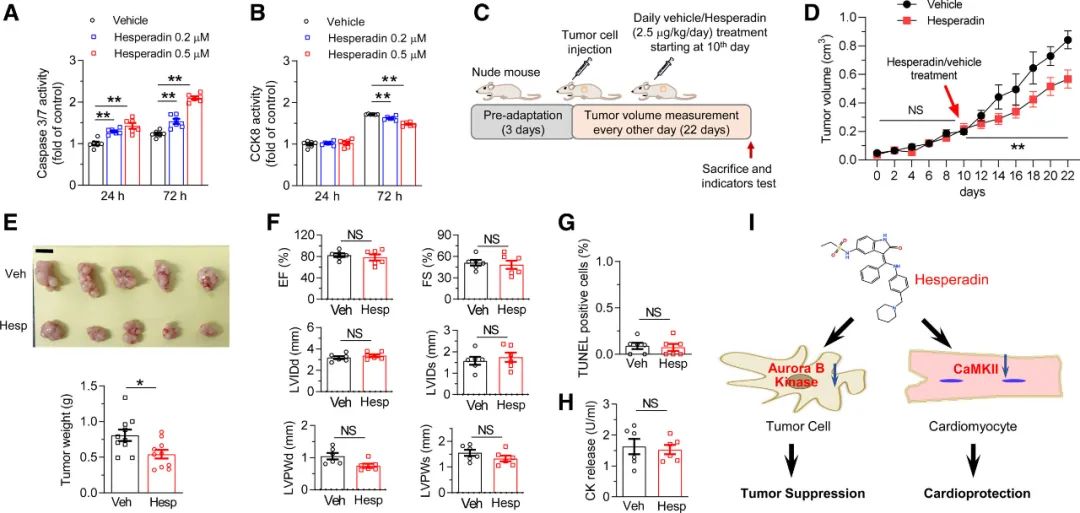
图8 Hesperadin能抑制肿瘤生长而不引起**损伤
综上,与其他生物药物靶点相比,激酶容易受到小分子的抑制。小分子疗法也**了许多优势,包括避免免疫反应、降低成本、以及能够到达细胞内等。本研究中,作者发现Hesperadin是CaMKII-δ的特异性小分子抑制剂,具有**保护和抗肿瘤的双重作用。这些发现不仅表明Hesperadin是临床治疗**I/R损伤和心力衰竭的一种有前景的先导化合物,而且为抗癌治疗引起的癌症和心血管疾病的联合治疗**了一种策略。

医影传声-为什么要做乳腺影像检查? 顾雅佳

如何读懂乳腺影像学检查报告? 汪登斌

基层麻醉主任对话主委一 邓小明 马虹 黄文起 陈向东 李翊堂 熊高钊

